Cada substância farmacêutica possui características químicas e físicas intrínsecas que devem ser consideradas antes do desenvolvimento de sua formulação farmacêutica. Entre essas características estão o tamanho da partícula, a área superficial, a solubilidade da droga, o pH, o coeficiente de partição, a taxa de dissolução, a forma física e a estabilidade. Todos esses fatores são discutidos abaixo, exceto o tamanho da partícula e a taxa de dissolução, que serão discutidos no próximo capítulo.
1.1. Tamanho e volume molecular
O tamanho e o volume molecular têm implicações importantes para a absorção do fármaco. As junções fechadas podem bloquear a passagem até mesmo de moléculas relativamente pequenas, enquanto as junções comunicantes são mais frouxas. Moléculas de até 1.200 Da podem passar livremente entre as células; no entanto, moléculas maiores não podem passar pelas junções comunicantes. A difusão da droga em líquido simples é expressa pela equação de Stokes-Einstein:
(eq. 1)
D= RT/6πnr
Onde o D é a difusão de drogas, o R é a constante do gás = 8,313 JK-1 mol-1, T é a temperatura (Kelvin), n é a viscosidade do solvente e r é o raio solvatado do soluto em difusão.
Como o volume (V) = (4/3) πr3, a equação acima sugere que a difusividade do fármaco é inversamente proporcional ao volume molecular. O volume molecular depende do peso molecular, conformação e conteúdo de heteroátomos. Moléculas com uma conformação compacta terão um volume molecular menor e, portanto, uma maior difusividade. A difusão e a permeabilidade da monocamada endotelial às moléculas diminuíram com o aumento do peso molecular.
Um fármaco deve se difundir através de uma variedade de membranas biológicas após a administração no corpo. Além disso, as drogas em muitos sistemas de liberação controlada devem se difundir através de uma membrana ou matriz controladora de velocidade. A capacidade de um fármaco se difundir através das membranas é uma função de seu tamanho e volume molecular. Para drogas com peso molecular maior que 500, a difusão em muitas matrizes poliméricas é muito pequena. Lipinski criou a chamada Regra dos 5, que se refere às propriedades das moléculas semelhantes às drogas. Afirma que a má absorção oral é mais provável quando a molécula da droga tem:
- Mais de cinco doadores de ligações de hidrogênio (grupos –OH ou grupos –NH).
- Um peso molecular superior a 800.
- Um log P > 5.
- Mais de 10H-bond aceitadores.
No entanto, esta regra não é aplicável aos compostos que são substratos para transportadores.
1.2. Solubilidade e pH do fármaco
A atividade farmacológica depende da solubilização de um fármaco em fluidos fisiológicos. Portanto, um fármaco deve possuir alguma solubilidade aquosa para absorção sistêmica e resposta terapêutica. A solubilidade aquosa aumentada pode ser alcançada pela formação de sais ou ésteres, por complexação química, pela redução do tamanho de partícula da droga (isto é, micronização) ou pela criação de um sólido amorfo. Um dos fatores mais importantes no processo de formulação é o pH, pois afeta a solubilidade e a estabilidade de compostos fracamente ácidos ou básicos. Alterações no pH podem levar à ionização ou formação de sal. O ajuste do pH é freqüentemente usado para aumentar a solubilidade de drogas ionizáveis, porque as espécies moleculares ionizadas têm maior solubilidade em água do que suas espécies neutras. A solubilidade total é a função da solubilidade intrínseca e a diferença entre o pKa da molécula e o pH da solução. A solubilidade intrínseca é a solubilidade das espécies neutras. Os ácidos fracos podem ser solubilizados em pHs abaixo de seu pKa ácido, enquanto as bases fracas podem ser solubilizadas em pHs acima de seu pKa básico. Para cada unidade de pH distante do pKa, a solubilidade ácido-base fraca aumenta 10 vezes. Assim, a solubilidade pode ser alcançada desde que o pH da formulação esteja pelo menos 3 unidades distante do pKa. Ajustar o pH da solução é o método mais simples e comum para aumentar a solubilidade em água em produtos injetáveis.
Ao contrário de um ácido ou base fraca, a solubilidade de um ácido ou base forte é menos afetada pelo pH. As drogas sem grupos ionizáveis são muitas vezes solubilizadas pela combinação de uma solução aquosa e solvente orgânico/surfactante solúvel em água. Freqüentemente, um soluto é mais solúvel em uma mistura de solventes do que em um solvente sozinho. Esse fenômeno é conhecido como cosolvência, e os solventes que, em combinação, aumentam a solubilidade do soluto são chamados de cossolventes. A adição de um co-solvente pode aumentar a solubilidade de moléculas hidrofóbicas pela redução da constante dielétrica, que é uma medida da influência de um meio na energia necessária para separar dois corpos de cargas opostas. Alguns dos co-solventes comumente usados em formulações farmacêuticas incluem álcool etílico, glicerina, sorbitol, propilenoglicol e polietilenoglicóis (PEGs). Polietilenoglicol 300 ou 400, propilenoglicol, glicerina, dimetilacetamida (DMA), N-metil 2-pirrolidona (NMP), dimetilsulfóxido (DMSO), Cremophor e polissorbato 80 são frequentemente usados para solubilização de drogas que não possuem grupos ionizáveis. Conforme mostrado na Figura 3.2, a solubilidade do fenobarbital é, por exemplo, significativamente aumentada em uma mistura de água, álcool e glicerina em comparação com um desses solventes sozinho. No entanto, o uso de co-solventes muitas vezes leva à precipitação do fármaco na diluição durante a administração da solução do fármaco no corpo, resultando em dor ou dano tecidual.
Excipientes que solubilizam uma molécula por meio de interações específicas, como a complexação com uma molécula de fármaco de maneira não covalente, diminuem o potencial químico das moléculas em solução. Essas interações não covalentes que aumentam a solubilidade são a base do fenômeno que semelhante dissolve semelhante e inclui forças de van der Waals, ligações de hidrogênio, interações dipolo-dipolo, íon-dipolo e, em certos casos, interações eletromagnéticas favoráveis. Os solutos se dissolvem melhor em solventes de polaridade similar. Portanto, para dissolver um composto altamente polar ou iônico, deve-se usar um solvente altamente polar ou com alta constante dielétrica. Ao contrário, para dissolver um fármaco apolar, deve-se usar um solvente relativamente apolar ou com baixa constante dielétrica.
A solubilidade do fármaco também pode ser aumentada pela alteração de sua estrutura; esta é uma base para o uso de pró-drogas. Um pró-fármaco é um fármaco que é terapeuticamente inativo quando administrado, mas é ativado no corpo por processamento químico ou enzimático. A adição de grupos polares, como ácidos carboxílicos, cetonas e aminas, pode aumentar a solubilidade aquosa por aumentar a ligação de hidrogênio e a interação dipolo-dipolo entre a molécula do fármaco e as moléculas de água. A Tabela 1 lista diferentes substituintes que terão influência significativa na hidrossolubilidade dos medicamentos. Os substituintes podem ser classificados como hidrofóbicos ou hidrofílicos, dependendo de sua polaridade. A posição dos substituintes na molécula também pode influenciar seu efeito.
A solubilidade do fármaco também pode ser aumentada pela alteração de sua estrutura; esta é uma base para o uso de pró-drogas. Um pró-fármaco é um fármaco que é terapeuticamente inativo quando administrado, mas é ativado no corpo por processamento químico ou enzimático. A adição de grupos polares, como ácidos carboxílicos, cetonas e aminas, pode aumentar a solubilidade aquosa por aumentar a ligação de hidrogênio e a interação dipolo-dipolo entre a molécula do fármaco e as moléculas de água. A Tabela 1 lista diferentes substituintes que terão influência significativa na hidrossolubilidade dos medicamentos. Os substituintes podem ser classificados como hidrofóbicos ou hidrofílicos, dependendo de sua polaridade. A posição dos substituintes na molécula também pode influenciar seu efeito.
Tabela 1. Solubilidade em água de diferentes grupos substituintes
| Grupos substituintes hidrofóbicos
–CH3 –CH2– –Cl,–Br –N(CH3)2 –SCH3 –OCH2CH3 |
| Grupos substituintes hidrofílicos
–OCH3 –NO2 –CHO –COOH –COO– –NH2 –NH3+ –OH |
1.3. Lipofilicidade e coeficiente de partição
A partição é a capacidade de um composto se distribuir em dois líquidos imiscíveis. Quando um ácido fraco ou um fármaco básico é adicionado a dois líquidos imiscíveis, algum fármaco vai para a fase apolar e outro vai para a fase aquosa. Como semelhante dissolve semelhante, as espécies apolares migram (partições) para a camada apolar e as espécies polares migram para a camada aquosa polar.
Para produzir uma resposta farmacológica, uma molécula de droga deve primeiro atravessar uma membrana biológica, que atua como uma barreira lipofílica para muitas drogas. Como a difusão passiva é o mecanismo predominante pelo qual muitas drogas são transportadas, a natureza lipofílica das moléculas é importante. O coeficiente de partição de um fármaco é uma medida de sua distribuição em um sistema de fase lipofílica-hidrófila e indica a capacidade do fármaco de penetrar em sistemas biológicos multifásicos. O coeficiente de partição octanol-água é comumente usado no desenvolvimento de formulações e é definido como:
(eq. 2)
P=(Concentração da droga em octanol ou fase apolar)/(Concentração do fármaco em água ou fase polar)
Para uma droga ionizável, a seguinte equação é aplicável:
(eq. 2)
Nesta equação, α é igual ao grau de ionização. A concentração na fase aquosa é estimada por um ensaio analítico, e a concentração em octanol ou outras fases orgânicas é deduzida subtraindo a quantidade aquosa da quantidade total colocada nos solventes. O coeficiente de partição pode ser usado para extração de drogas de plantas ou fluidos biológicos, absorção de drogas de formas de dosagem e recuperação de antibióticos do caldo de fermentação.
O logaritmo do coeficiente de partição (P) é conhecido como log P. Log P é uma medida de lipofilicidade e é amplamente utilizado, uma vez que muitos eventos farmacêuticos e biológicos dependem de características lipofílicas. Freqüentemente, o log P de um composto é citado. Para um determinado medicamento:
- Se log P = 0, há distribuição igual da droga em ambas as fases.
- Se log P > 0, o fármaco é lipossolúvel.
- Se log P < 0, a droga é solúvel em água.
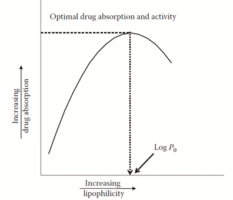
Figura 1. Relação entre a absorção do fármaco e o log P. A diminuição da absorção do fármaco além de um determinado valor de log P deve-se provavelmente à sua ligação às proteínas plasmáticas, à redução dos níveis do fármaco livre ou à sua ligação a locais estra
Em geral, quanto maior o log P, maior a afinidade pelas membranas lipídicas e, portanto, mais rapidamente o fármaco passa através da membrana por difusão passiva. No entanto, existe uma relação parabólica entre log P e a atividade da droga quando a porcentagem de absorção da droga é plotada contra os valores de log P. A natureza parabólica da bioatividade e dos valores de log P se deve ao fato de que drogas com altos valores de log P, ligação a proteínas, baixa solubilidade e ligação a locais estranhos fazem com que tenham uma bioatividade mais baixa. A diminuição da atividade é devida à limitação da solubilidade além de um certo valor de log P. Se um fármaco for muito lipofílico, ele permanecerá na membrana lipídica e não será particionado novamente no ambiente aquoso subjacente. Por outro lado, compostos muito polares com valores de log P muito altos não são suficientemente lipofílicos para serem capazes de passar através das barreiras lipídicas da membrana.
1.4. Polimorfismo
A capacidade de uma substância existir em mais de uma forma de estado sólido é conhecida como polimorfismo, e as diferentes formas cristalinas são chamadas de polimorfos. Se a mudança de um polimorfo para outro for reversível, o processo é enantiotrópico. No entanto, se a transição de um metaestável para um polimorfo estável for unidirecional, o sistema é monotrópico. As formas polimórficas podem exibir diferenças detectáveis em algumas ou todas as seguintes propriedades: ponto de fusão, taxa de dissolução, solubilidade e estabilidade. As substâncias medicamentosas podem ser amorfas (ou seja, sem arranjos regulares de rede molecular), cristalinas (mais orientadas ou alinhadas), polimórficas, anidras ou solvatadas. Uma forma importante na formulação é a forma cristalina ou amorfa do fármaco. Muitas substâncias medicamentosas podem existir em mais de uma forma cristalina, com diferentes arranjos de rede. Esta propriedade é denominada polimorfismo. As drogas podem passar de uma forma polimórfica metaestável para uma forma polimórfica mais estável. Vários medicamentos são conhecidos em diferentes formas polimórficas (por exemplo, cortisona e prednisolona). As formas polimórficas geralmente exibem diferentes propriedades físico-químicas, incluindo ponto de fusão e solubilidade, que podem afetar a taxa de dissolução e, portanto, a extensão de sua absorção. A forma amorfa de um composto é sempre mais solúvel do que a forma cristalina correspondente. Alterações nas características do cristal podem influenciar a biodisponibilidade e a estabilidade e, portanto, podem ter implicações importantes para o design da forma farmacêutica. Por exemplo, a insulina exibe um grau diferente de atividade, dependendo do seu estado. A forma amorfa da insulina é rapidamente absorvida e tem ação de curta duração, enquanto o produto cristalino grande é absorvido mais lentamente e tem ação de duração mais longa.
1.5. Estabilidade
A estabilidade química e física de um fármaco isoladamente e quando combinada com componentes de formulação é crítica para a preparação de um produto farmacêutico bem-sucedido. Drogas contendo um dos seguintes grupos funcionais podem sofrer degradação hidrolítica: éster, amida, lactose, lactama, imida ou carbamato. Drogas que contêm ligações éster incluem ácido acetilsalicílico, fisostigmina, metildopa, tetracaína e procaína. Por exemplo, a hidrólise do ácido acetilsalicílico (comercialmente conhecido como aspirina). A aspirina é hidrolisada em ácido salicílico e ácido acético.
Nitrazepam, clordiazepóxido, penicilinas e cefalosporinas também são suscetíveis à hidrólise. Vários métodos estão disponíveis para estabilizar soluções de drogas que são suscetíveis à hidrólise. Por exemplo, proteção contra umidade na formulação, processamento e embalagem pode prevenir a decomposição. A suspensão de medicamentos em solventes não aquosos, como álcool, glicerina ou propilenoglicol, também pode reduzir a hidrólise.
Após a hidrólise, a oxidação é a próxima via mais comum para a degradação do fármaco. Drogas que sofrem degradação oxidativa incluem morfina, dopamina, adrenalina, esteróides, antibióticos e vitaminas. A oxidação pode ser minimizada pelo armazenamento em condições anaeróbicas. Como é muito difícil remover todo o oxigênio de um recipiente, os antioxidantes são frequentemente adicionados às formulações para evitar a oxidação.
Os excipientes usados para preparar uma dosagem sólida também podem afetar a eficácia do medicamento.
estabilidade, possivelmente aumentando o teor de umidade da preparação. Excipientes, como amido e povidona, possuem teores de água muito altos.
A povidona contém cerca de 28% de umidade de equilíbrio a 75% de umidade relativa. No entanto, o efeito desse alto teor de umidade na estabilidade de uma droga dependerá de quão fortemente ele está ligado e se a umidade pode entrar em contato com a droga. Os efeitos dos excipientes de comprimidos na decomposição de drogas são amplamente relatados na literatura. Por exemplo, o trissilicato de magnésio é conhecido por causar aumento da hidrólise da aspirina no comprimido devido ao seu alto teor de umidade.
1.6. Pka ou constante de dissociação
Muitas substâncias farmacêuticas são ácidos fracos ou bases fracas e, portanto, sofrem um fenômeno conhecido como dissociação quando dissolvidas em meio líquido. Se esta dissociação envolve uma separação de cargas, então há uma mudança na distribuição de carga elétrica na espécie e uma separação em duas ou mais partículas carregadas, ou ionização. A extensão da ionização de um fármaco tem um efeito importante na formulação e nos perfis farmacocinéticos do fármaco. A extensão da dissociação ou ionização depende do pH do meio que contém o fármaco. A Tabela 3.4 lista os pHs normais de alguns órgãos e fluidos corporais, que são usados na previsão da porcentagem de ionização de drogas in vivo. Em uma formulação, muitas vezes, o veículo é ajustado a um determinado pH para obter um certo nível de ionização do fármaco para solubilidade e estabilidade. A extensão da ionização de um fármaco tem forte efeito sobre sua extensão de absorção, distribuição e eliminação.
Os ácidos tendem a doar prótons para um sistema com pH maior que 7, e as bases tendem a aceitar prótons quando adicionadas ao sistema ácido (ou seja, em pH < 7). Muitas drogas são ácidos ou bases fracos e, portanto, existem tanto na forma não ionizada quanto na ionizada; a proporção dessas duas formas varia com o pH. A fração de uma droga que é ionizada em solução é dada pela constante de dissociação (Ka) da droga. Essas constantes de dissociação são convenientemente expressas em termos de valores de pKa para drogas ácidas e básicas.
Tanto para ácidos quanto para bases fracas, as espécies ionizadas têm solubilidade muito baixa e são virtualmente incapazes de permear a membrana, exceto onde existem mecanismos específicos de transporte. A lipossolubilidade dos fármacos não carregados dependerá das propriedades físico-químicas do fármaco.
Proteínas e peptídeos contêm grupos ácidos (−COOH) e básicos (−NH2). Os valores de pKa de grupos ionizáveis em proteínas e peptídeos podem ser significativamente diferentes daqueles dos grupos correspondentes quando eles são isolados em solução. Portanto, esses compostos são frequentemente referidos como de natureza anfotérica. O pH de uma solução determina a carga líquida na molécula e, finalmente, a solubilidade. Como a água é um solvente polar e as espécies iônicas são mais solúveis em água do que as não iônicas, um ácido conjugado e uma base conjugada são geralmente mais solúveis em água do que a base livre correspondente ou o ácido livre.
1.7. Grau de ionização e teoria de partição de pH
Para que um fármaco atravesse uma barreira de membrana, normalmente deve ser lipossolúvel para entrar nas membranas biológicas. As formas ionizadas de drogas ácidas e básicas têm baixos coeficientes de partição lipídio:água em comparação com os coeficientes das moléculas não ionizadas correspondentes. As membranas lipídicas são preferencialmente permeáveis às últimas espécies. Assim, um aumento na fração de um fármaco que está sindicalizado aumentará a taxa de transporte do fármaco através da membrana lipídica. Esse fenômeno pode ser explicado pela teoria da partição do pH, que afirma que os fármacos são absorvidos das membranas biológicas por difusão passiva, dependendo da fração da forma não ionizada do fármaco no pH dessa membrana biológica. Com base na equação de Henderson-Hasselbalch, o grau de ionização de uma droga dependerá tanto de seu valor de pKa quanto do pH da solução.
O trato gastrointestinal (GI) atua como uma barreira lipofílica e, portanto, as drogas ionizadas, que serão mais hidrofílicas, terão um transporte de membrana mínimo em comparação com a forma não ionizada da droga. O pH da solução afetará o coeficiente de partição geral de uma substância ionizável. O pI da molécula é o pH no qual existe uma mistura 50:50 de formas ácido-base conjugadas. A forma ácida conjugada predominará em um pH menor que o pKa, e a forma básica conjugada estará presente em um pH maior que o pKa.
1.7.1. Limitações da teoria da partição de pH
Embora a teoria da partição de pH seja útil, muitas vezes ela não é verdadeira. Por exemplo, a maioria dos ácidos fracos é bem absorvida no intestino delgado, o que é contrário à previsão da hipótese de partição do pH. Da mesma forma, os compostos de amônio quaternário são ionizados em todos os pHs, mas são facilmente absorvidos pelo trato GI. Essas discrepâncias surgem porque a teoria da partição de pH não leva em consideração o seguinte:
- O intestino delgado tem uma grande área de superfície epitelial para que ocorra a absorção do fármaco. Essa grande área epitelial resulta da mucosa, vilosidades e microvilosidades. A grande área de superfície da mucosa compensa os efeitos de ionização.
- As drogas têm um tempo de residência relativamente longo no intestino delgado,
- que também compensa os efeitos de ionização.
- Drogas carregadas, como compostos de amônio quaternário e tetraciclinas, podem interagir com íons orgânicos de carga oposta, resultando em uma espécie neutra, que é absorvível.
- Algumas drogas são absorvidas por vias ativas.